Page 1 of 14
SU19.1-2 | Cleft Lip and Palate — SDL Guide
Learning Objectives
- Describe the embryology, classification and aetiology of cleft lip and palate (SU19.1).
- Describe the principles of reconstruction of cleft lip and palate, including the timing and sequence of repair (SU19.2).
INSTRUCTIONS
Cleft lip and palate is the commonest congenital anomaly of the face, and few conditions illustrate so clearly how a single embryological event ripples through feeding, hearing, speech, dental development and a family's sense of well-being. The surgical task is not merely to close a gap in tissue but to restore form and function on a biological timetable: the lip is repaired in infancy to allow normal appearance and bonding, and the palate is repaired before speech develops. This module connects the embryology and classification of the cleft to a structured multidisciplinary assessment and the principles of staged reconstruction.
References
- Bailey & Love's Short Practice of Surgery, Cleft Lip and Palate / Plastic and Reconstructive Surgery (textbook)
- SRB's Manual of Surgery, Cleft Lip and Cleft Palate (textbook)
- Sabiston Textbook of Surgery, Plastic Surgery — Congenital Anomalies of the Face (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A mother brings her newborn son to the clinic on the second day of life. She is anxious, exhausted and quietly distressed: there is a visible gap in her baby's upper lip running up to one nostril, and feeding has been a battle through the night because milk keeps escaping from his nose. In the same waiting area sits another family whose three-year-old daughter, born with a cleft of the palate that was repaired in infancy, has been referred because her speech is hard to understand and she has had repeated ear infections. The same fundamental anomaly — a failure of the facial and palatal structures to fuse in the womb — has produced two very different stories separated by three years and several operations. Understanding the embryology beneath that visible gap, and the orderly timetable of repair that follows, is what turns a frightening diagnosis into a manageable, treatable journey.
WHY THIS MATTERS
Cleft lip and palate is the most common congenital anomaly of the orofacial region, affecting roughly one in 700 to 1000 live births, so any doctor working with children, mothers or surgical referrals will meet it. It matters far beyond the cosmetic gap a layperson sees: an unrepaired cleft impairs feeding and nutrition in the newborn, an unrepaired palate causes nasal regurgitation, recurrent middle-ear disease and disordered speech, and the whole condition carries a real psychosocial burden for the child and family. For a final-year student, cleft care is also a model of multidisciplinary surgery — the surgeon works alongside paediatrician, neonatologist, anaesthetist, orthodontist, speech therapist, audiologist and psychologist on a shared timetable. Knowing the classification lets you describe a cleft precisely, and knowing the principles and timing of reconstruction lets you counsel a family on what will happen and when.
RECALL
Recall some foundations before we build on them. From embryology: the face forms in the first trimester by the fusion of several processes around the developing mouth — the frontonasal process gives rise to the medial and lateral nasal processes, and the paired maxillary processes grow forwards from the first pharyngeal arch. The upper lip and the primary palate (the part of the palate in front of the incisive foramen) are formed by the fusion of the medial nasal and maxillary processes, while the secondary palate (behind the incisive foramen) forms later by the fusion of the two palatal shelves in the midline. A failure of fusion at any of these points leaves a cleft. From physiology, remember that the newborn feeds by creating an intra-oral seal and suction; a cleft destroys that seal, which is why feeding difficulty is an early and important problem. Hold on to one anatomical landmark in particular — the incisive foramen — because it is the dividing line that organises the entire classification of clefts.
The Child Born with a Cleft
A cleft lip is usually obvious at birth or even on antenatal ultrasound: there is a visible gap in the upper lip, on one side of the midline (unilateral, the commoner pattern) or on both (bilateral), and it may extend up into the floor of the nostril, flattening and distorting the nose. A cleft palate may be less immediately obvious, particularly an isolated cleft of the soft palate, and is sometimes found only when a careful examination of the mouth is performed or when feeding problems prompt a closer look. The earliest and most pressing functional problem is feeding difficulty: because the cleft prevents the infant from generating the negative intra-oral pressure needed to suckle, milk escapes, feeds are prolonged and ineffective, milk regurgitates through the nose, and the baby is at risk of poor weight gain and aspiration. Over the following months and years, an unrepaired or repaired cleft palate brings further functional consequences that the family will notice: recurrent middle-ear infections and hearing loss, because the cleft disrupts the muscles that open the Eustachian tube and ventilate the middle ear; speech difficulty, characteristically a hypernasal, hard-to-understand voice from velopharyngeal incompetence; and dental and orthodontic problems where the cleft crosses the alveolus. Beyond function, the visible deformity carries a genuine psychosocial impact on the child and a heavy emotional load on the parents at the moment of birth. A cleft may also be one feature of a wider syndrome — for example the Pierre Robin sequence, in which a small lower jaw and a backward-displaced tongue accompany the cleft palate and can threaten the airway — so the first clinical task is to assess the whole child, secure feeding and the airway, and identify any associated anomalies before planning any operation.
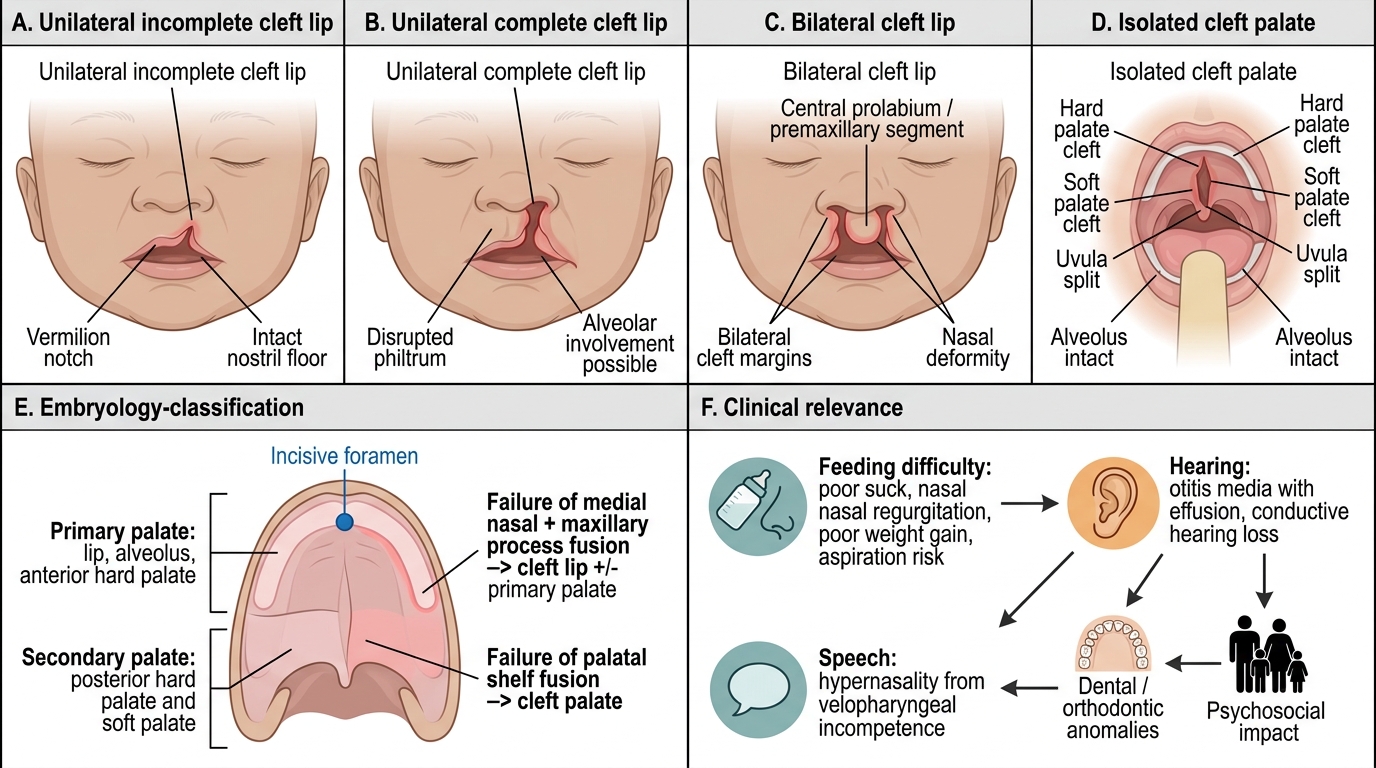
Spectrum and Classification of Cleft Lip and Palate
- Feeding difficulty: loss of intra-oral seal → poor suck, nasal regurgitation, poor weight gain, aspiration risk.
- Hearing: recurrent otitis media with effusion and conductive hearing loss (palatal muscle dysfunction).
- Speech: hypernasal, unintelligible speech from velopharyngeal incompetence.
- Dental/orthodontic and psychosocial: alveolar/dental anomalies; significant impact on child and family.
Embryology, Classification and Aetiology
The cleft is best understood as a failure of the normal embryological fusion you recalled earlier, and the incisive foramen is the anatomical key that organises the whole classification. The lip and the primary palate — the alveolus and the hard palate in front of the incisive foramen — are formed by fusion of the medial nasal and maxillary processes; a failure here produces a cleft lip with or without a cleft of the primary palate. The secondary palate — the hard and soft palate behind the incisive foramen — forms by the later fusion of the two palatal shelves; a failure here produces a cleft palate. This embryology explains why clefts are described along three independent axes. By site, a cleft may involve the lip alone, the palate alone, or both lip and palate together. By laterality, a cleft lip may be unilateral (more common, and more often on the left) or bilateral. By completeness, a cleft may be incomplete (for example a notch in the lip that does not reach the nostril) or complete (extending the full height of the lip into the nostril floor, or the full length of the palate). Aetiologically, most clefts are non-syndromic and multifactorial, arising from an interplay of genetic susceptibility and environmental factors such as maternal smoking, alcohol, folate deficiency, certain anticonvulsants and maternal diabetes. A minority are syndromic, occurring as part of a recognised syndrome such as the Pierre Robin sequence (micrognathia, glossoptosis and cleft palate) or Van der Woude syndrome; this distinction matters because syndromic children need a broader work-up and may have airway or other anomalies. Epidemiologically, combined cleft lip and palate and isolated cleft lip are commoner in boys, whereas isolated cleft palate is commoner in girls.
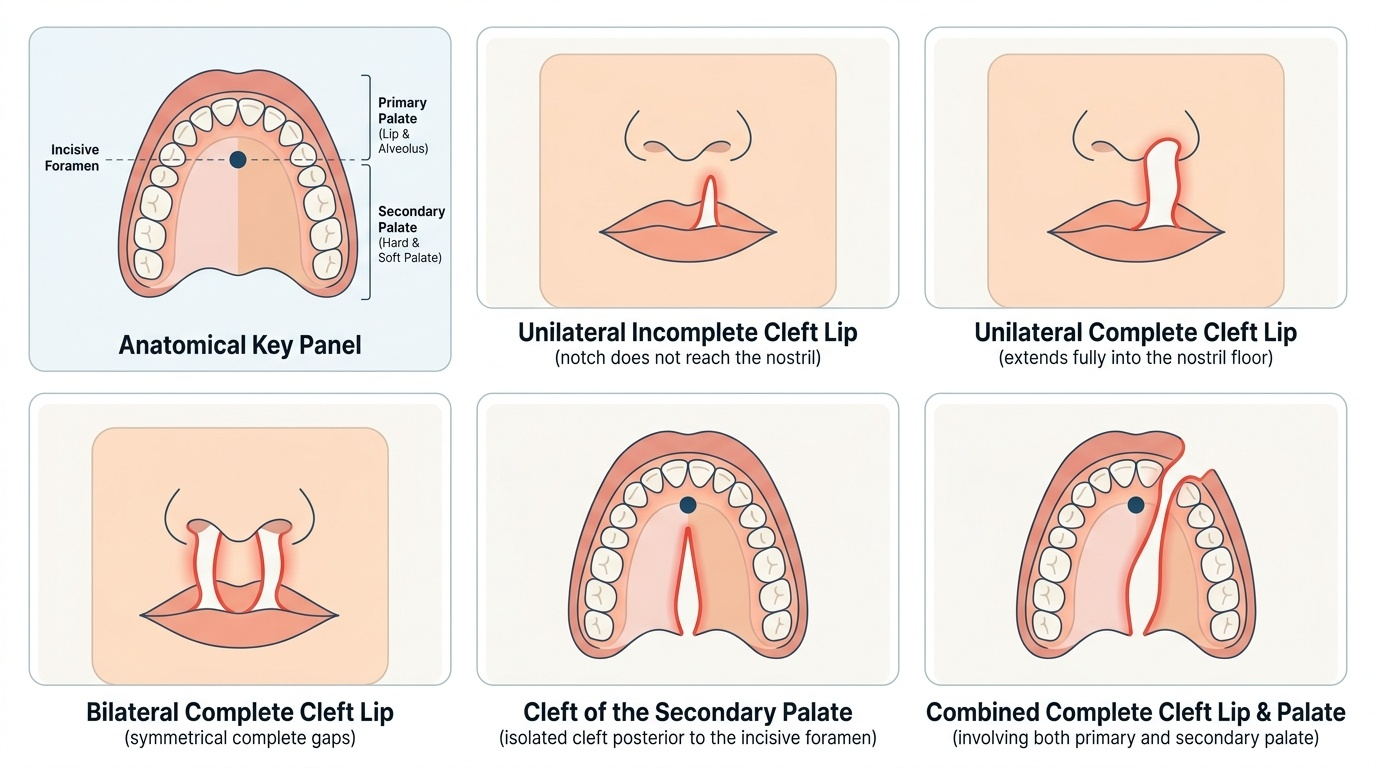
Provided image
- Site: cleft lip alone; cleft palate alone; combined cleft lip and palate.
- Laterality: unilateral (commoner, often left) vs bilateral.
- Completeness: incomplete vs complete.
- Aetiology: non-syndromic multifactorial (genetic + environmental) vs syndromic (e.g. Pierre Robin sequence).
SELF-CHECK
Which anatomical landmark divides the primary palate from the secondary palate and therefore organises the classification of cleft palate?
A. The hard-soft palate junction
B. The incisive foramen
C. The posterior nasal spine
D. The greater palatine foramen
Reveal Answer
Answer: B. The incisive foramen
The incisive foramen is the embryological and anatomical dividing line: structures anterior to it (lip and alveolus/anterior hard palate) form the PRIMARY palate from the medial nasal and maxillary processes, while structures posterior to it form the SECONDARY palate from fusion of the palatal shelves. Clefts are classified relative to this landmark.
Assessment and the Multidisciplinary Work-up
Assessment of a child with a cleft begins not with the surgery but with the whole baby, and it is a fundamentally multidisciplinary process spread over years rather than a single clinic visit. The immediate priorities at birth are to secure the airway — especially in the Pierre Robin sequence, where the small jaw and posteriorly placed tongue can cause obstruction that may need positioning, a nasopharyngeal airway or, rarely, more — and to establish feeding, with specialised cleft feeding teats and bottles, upright positioning and support from a feeding specialist so that the baby grows and is fit for surgery. A careful clinical examination documents the cleft precisely along the three axes (site, laterality, completeness) and, critically, examines the rest of the child for associated anomalies that would suggest a syndrome, prompting paediatric and genetic evaluation. Because the cleft affects the middle ear, every child needs hearing assessment and ENT review for the otitis media with effusion that is almost universal, often managed with grommets. As the child grows, speech and language assessment by a speech therapist tracks velopharyngeal function and guides whether secondary speech surgery will be needed, while orthodontic and dental assessment plans for the alveolar and dental anomalies. The team therefore typically includes a plastic or maxillofacial surgeon, paediatrician/neonatologist, anaesthetist, ENT surgeon, audiologist, orthodontist, speech and language therapist, specialist nurse and psychologist, all working to a shared protocol. Routine imaging is not required to diagnose the cleft itself, which is a clinical diagnosis, but investigations are directed at the work-up of associated problems and at pre-operative fitness — confirming adequate weight gain and a safe haemoglobin before the baby is taken to theatre.
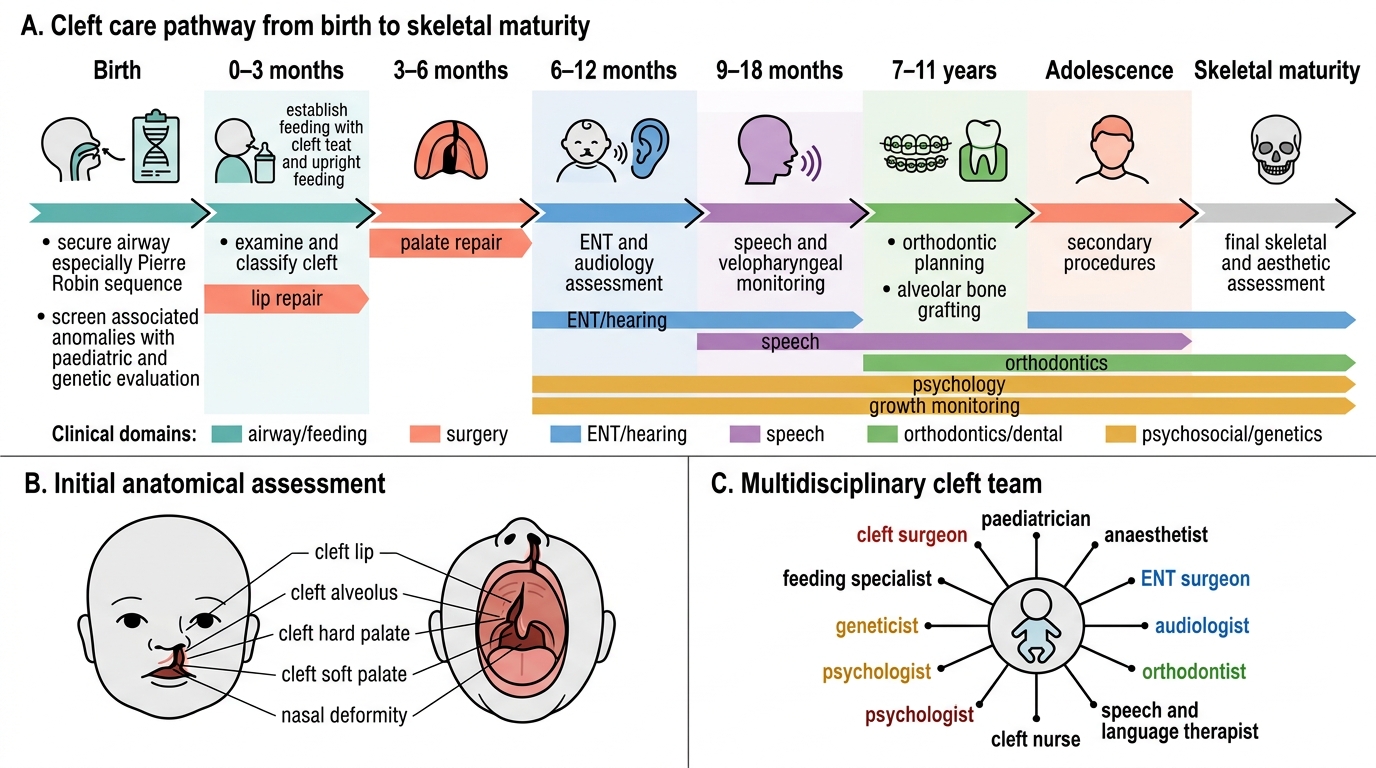
Multidisciplinary Cleft Care Pathway
- First: secure airway (esp. Pierre Robin) and establish feeding (cleft teats, upright feeding, feeding specialist) to ensure growth.
- Examine and classify: document site/laterality/completeness; screen for associated anomalies → paediatric + genetic evaluation.
- ENT/audiology: assess and manage otitis media with effusion and conductive hearing loss.
- Speech and orthodontics: track velopharyngeal function and plan alveolar/dental care over time.
- Team: surgeon, paediatrician, anaesthetist, ENT, audiologist, orthodontist, speech therapist, nurse, psychologist.